How Do You Know if a Reaction Is Closed or Open Transition State
Stereoselective Synthesis |
|---|
Stereoselectivity refers to the preferential formation in a chemical reaction of one production stereoisomer (enantiomer or diastereomer) over another, as a upshot of inherent reaction specificity, or the influence of chiral features in the substrate, reagent, catalyst or surround. The more specific terms enantioselectivity and diastereoselectivity are commonly used in appropriate situations. When this selectivity results in the formation of an excess of one enantiomer over the other from an achiral or racemic substrate it is sometimes called asymmetric induction.
Whatsoever reaction which creates a new stereogenic center may proceed in a stereoselective style. The reduction of 3-hexyne to trans-3-hexene by sodium in ammonia, or to cis-3-hexene by Lindlar catalytic hydrogenation are examples. Since the substrate, reagents and products are all achiral, this diastereospecificity lies in the nature of the reactions themselves.
Addition to Carbon-Carbon Double Bonds
Hydroboration
Hydroboration of the prochiral alkene one-phenylcyclopentene, followed by peroxide oxidation, yields trans-two-phenylcyclopentanol equally a racemic mixture. 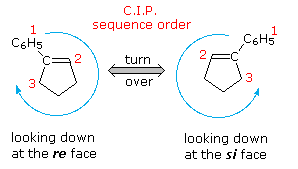 Two new stereogenic centers have been created in this diastereoselective reaction, but since reaction takes identify equally at both faces of the double bond, a l:fifty mixture of enantiomers is obtained (equation one of the following diagram). This double bond, and others having three unlike substituents at one carbon, take enantiotopic faces in the same sense that bromochloromethane has a pair of enantiotopic hydrogens. These enantiotopic faces are shown in the diagram on the right. The three different substituents are ranked by the Cahn-Ingold-Prelog sequence rules, and the double bail is viewed from a point above or below its plane. The sequence guild is then adamant to be a right or left plough (calorie-free blueish arrow), and the viewing face is given the designation re or si, corresponding to the turn. An addition of a given reagent at the re face volition necessarily give the enantiomer of the same addition at the si face, provided the new substituent is non identical to one of the three original groups. Clearly, to make hydroboration an enantioselective reaction, the rate of reaction at 1 of the faces must be increased over that at the other face. 1 way of accomplishing this is apply an enantiomerically pure chiral alkyl borane reagent for the hydroboration step. The oxidation step is known to proceed with memory of configuration. Several reagents of this kind have been prepared, ane useful i being monoisopinocampheylborane (IpcBH2), prepared as shown from the abundant terpene α-pinene. When used for the hydroboration of 1-phenylcyclopentene, the 1R,2S enantiomer is the sole product (equation 2 below). The respective diisopinocampheylborane (Ipc2BH) has also been used in similar enantioselective syntheses, just in general the enantiomeric purity of products from reactions with either reagent is seldom as high every bit the case given here.
Two new stereogenic centers have been created in this diastereoselective reaction, but since reaction takes identify equally at both faces of the double bond, a l:fifty mixture of enantiomers is obtained (equation one of the following diagram). This double bond, and others having three unlike substituents at one carbon, take enantiotopic faces in the same sense that bromochloromethane has a pair of enantiotopic hydrogens. These enantiotopic faces are shown in the diagram on the right. The three different substituents are ranked by the Cahn-Ingold-Prelog sequence rules, and the double bail is viewed from a point above or below its plane. The sequence guild is then adamant to be a right or left plough (calorie-free blueish arrow), and the viewing face is given the designation re or si, corresponding to the turn. An addition of a given reagent at the re face volition necessarily give the enantiomer of the same addition at the si face, provided the new substituent is non identical to one of the three original groups. Clearly, to make hydroboration an enantioselective reaction, the rate of reaction at 1 of the faces must be increased over that at the other face. 1 way of accomplishing this is apply an enantiomerically pure chiral alkyl borane reagent for the hydroboration step. The oxidation step is known to proceed with memory of configuration. Several reagents of this kind have been prepared, ane useful i being monoisopinocampheylborane (IpcBH2), prepared as shown from the abundant terpene α-pinene. When used for the hydroboration of 1-phenylcyclopentene, the 1R,2S enantiomer is the sole product (equation 2 below). The respective diisopinocampheylborane (Ipc2BH) has also been used in similar enantioselective syntheses, just in general the enantiomeric purity of products from reactions with either reagent is seldom as high every bit the case given here.
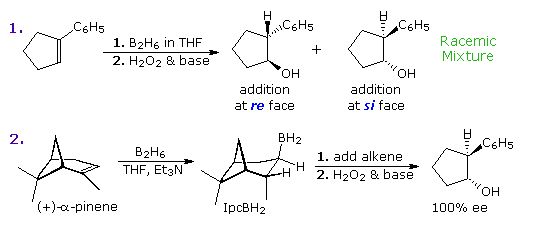
The ee percentage given in the last equation stands for enantiomeric excess, which is the mole fraction difference in composition of the enantiomers in a mixture. A racemic mixture has equal amounts of enantiomers, so the deviation is zero. If in that location is three times as much (+)-enantiomer as (-)-enantiomer the difference is [0.75 - 0.25 = 0.v] or 50% ee. A pure enantiomer has 100% ee. Differences in enantiomer concentration are normally measured by polarimetry, chiral phase chromatography or nmr with chiral shift reagents.
Epoxidation
Epoxidation of double bonds has proven to be an effective manner of introducing oxygen functionality at both carbon atoms. One or two new stereogenic centers are unremarkably created, often with excellent diastereoselectivity. This transformation is usually carried out by the activeness of a peracid (RCO3H), such as peracetic acrid or perbenzoic acid, in chloroform or methylene chloride solution. The configuration of double bond substituents (E or Z) is generally preserved, every bit shown in equation 1 beneath. Electron donating substituents on the double bond facilitate this reaction, and in the case of deactivation by conjugated electron withdrawing groups, reaction with alkylhydroperoxide anions oft achieves epoxidation by a cohabit improver-elimination pathway (equation ii). Steric hindrance commonly diverts epoxidation to the less hindered face up of a double bond; even so hydrogen bonding to a neighboring hydroxyl group may provide a more powerful orienting influence, as in equation 3. In this case note also the selective epoxidation of the more substituted double bond.
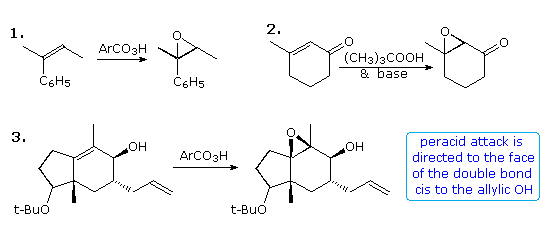
Alternative reaction paths for epoxidation may event in a different diastereoselectivity, as demonstrated above by clicking on the diagram. The trans-fused cycloalkene outlined by the low-cal green box in equation 4 may be epoxidized by a peracid or by base of operations treatment of a bromohydrin intermediate. The upper or β-face of the double bond is blocked past the athwart methyl group, then peracid epoxidation occurs at the other face (designated α). Addition of HOBr to the double bond is initiated by electrophilic bromine assail at the less-hindered α-face up, and since diaxial addition is favored stereoelectronically, the hydroxyl is bound to the β-face. An Intramolecular SDue northii reaction so forms the diastereomeric epoxide. A similar strategy permits diastereoselectivity to be achieved for acyclic alkenes, such as that shown in equation v. Here a nearby nucleophilic carboxyl group is positioned to reversibly open an iodonium intermediate, producing an iodolactone (fundamental structure). Since this is a reversible reaction, the more stable trans-disubstituted lactone is the major product. Base catalyzed opening of the lactone forms an alkoxide species that immediately displaces iodide to give the epoxide product. Notation that all the compounds in this equation are chiral and racemic.
Information technology is known that epoxidation of isolated alkenes by tert-butyl hydroperoxide may exist catalyzed by transition element catalysts, and that an allylic hydroxyl facilitates and facially directs the reaction. By clicking on the above diagram a second fourth dimension, examples of this reaction will be displayed, with equation 8 illustrating the neighboring group influence. Several features should be noted. Outset, the Z-configuration of the double bond is preserved in the epoxide products. Second, the allylic hydroxyl grouping exerts a modest facial diastereoselectivity on the reaction. For clarity the chiral carbinol group is drawn in its R-configuration, but the racemic alcohol would yield the same mixture of diastereomers as racemates.
Professor G. B. Sharpless, Scripps Inquiry Plant, has transformed this general epoxidation reaction into a powerful enantioselective procedure, past the add-on of a chiral tartrate ester ligand to a titanium alkoxide goad. This important constructed method is outlined in the post-obit diagram.
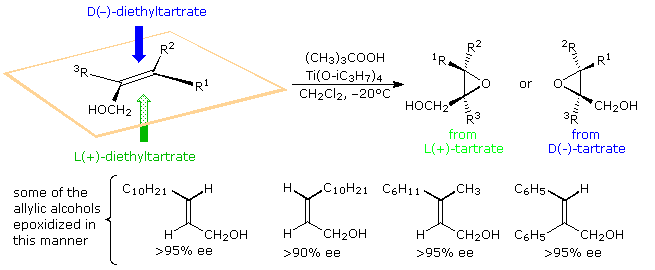
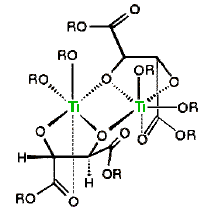 When mixed with one equivalent of diethyl tartrate, titanium tetraisopropoxide forms a dimeric complex with the loss of two isopropyl alcohol molecules. A proposed structure for this complex is shown on the right, with the titanium atoms colored green. Addition of tert-butylhydroperoxide and an allylic alcohol results in the displacement of ii more than isopropyl alcohols and the formation of a new reactive catalyst complex. A construction for this circuitous will as well be displayed on the right by clicking on the original structure. In this new cartoon the double bond of the allylic alcohol is colored blueish, and the peroxide oxygens, one of which becomes the epoxide oxygen, are colored ruby. A pinkish arrow indicates the bonding of this oxygen to a prochiral confront of the double bond. For primary allylic alcohols of the type shown above, Sharpless epoxidation achieves the remarkable conversion of an achiral substrate into a chiral production with high enantioselectivity.
When mixed with one equivalent of diethyl tartrate, titanium tetraisopropoxide forms a dimeric complex with the loss of two isopropyl alcohol molecules. A proposed structure for this complex is shown on the right, with the titanium atoms colored green. Addition of tert-butylhydroperoxide and an allylic alcohol results in the displacement of ii more than isopropyl alcohols and the formation of a new reactive catalyst complex. A construction for this circuitous will as well be displayed on the right by clicking on the original structure. In this new cartoon the double bond of the allylic alcohol is colored blueish, and the peroxide oxygens, one of which becomes the epoxide oxygen, are colored ruby. A pinkish arrow indicates the bonding of this oxygen to a prochiral confront of the double bond. For primary allylic alcohols of the type shown above, Sharpless epoxidation achieves the remarkable conversion of an achiral substrate into a chiral production with high enantioselectivity.
In the reaction of secondary allylic alcohols, where substituent R2 or Riii is an alkyl group (diagram on the right), the allylic alcohol substrate is chiral and the enantiomers react at dissimilar rates via diastereomeric transition states. For the racemic booze in which Rtwo (or R3) is a cyclohexyl group, the S-enantiomer reacts over 100 times faster than the R-enantiomer, presumably due to steric hindrance of R2. This charge per unit departure results in a kinetic resolution of this substrate. The S-enantiomer is converted to its erythro diastereomeric epoxide in 98:2 diastereospecificity, while the R-enantiomer is recovered unchanged in over 96% ee.
A model of the Sharpless goad may be examined by .
Other catalyst systems for enantioselective epoxidation, also as hydroxylation and hydrogenation of carbon-carbon double bonds, have been developed and are used in the manufacture of chiral intermediates. The 2001 Nobel Prize in chemistry was awarded to William S. Knowles, Ryoji Noyori, and Grand. Barry Sharpless for their seminal work in this of import field.
The previous examples illustrate an of import principle.
Enantioselectivity requires the presence of a chiral feature in a substrate, reagent or catalyst.
Diastereoselectivity does non require such chirality, only is normally produced past it.
Addition to Carbonyl Double Bonds
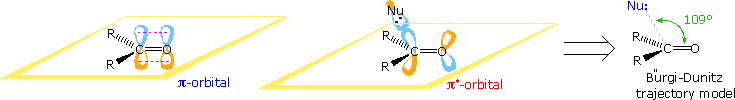
Stereoelectronic factors influence the improver of nucleophilic reagents to carbonyl groups, particularly aldehydes and ketones. In the post-obit diagram the essential molecular orbitals are drawn to the left of the arrow. Initial bonding with a nucleophile is believed to involve the empty antibonding π*-orbital. This explains the favored bonding alignment, known every bit the Bürgi-Dunitz trajectory.
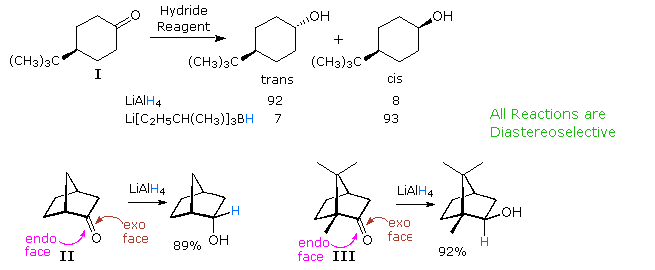
Nucleophilic improver reactions to aldehydes and ketones are probably the earliest and most actively studied examples of stereoselectivity. Selective reduction of 4-tert-butylcyclohexanone (I) to a x:ane mixture of trans- and cis-4-tert-butylcyclohexanol by LiAlHiv is an example of diastereoselectivity, reflecting a preference for hydride attack at the more hindered axial face up of the carbonyl group. This selectivity can be reversed by using a larger hydride reagent, such equally Li(sec-butyl)3BH; in which instance astringent hindrance to axial approach diverts the reaction to the equatorial face. The cis-alcohol is so establish to be the major product by over ten:i. Note that in these reactions both the reactants and products are achiral. A like influence of steric hindrance is seen in reductions of the bicyclo[ii.2.one]heptanones Ii and Iii (shown below). In compound II the exo face of the prochiral carbonyl group is less hindered than the endo confront. The hindrance is reversed in compound III, and the products from LiAlH4 reduction reflect this departure. The relatively fixed configurations of these circadian ketones permit local stereoelectronic and steric factors to influence reactivity in a well-defined style.
Models for Add-on to Acyclic Substrates
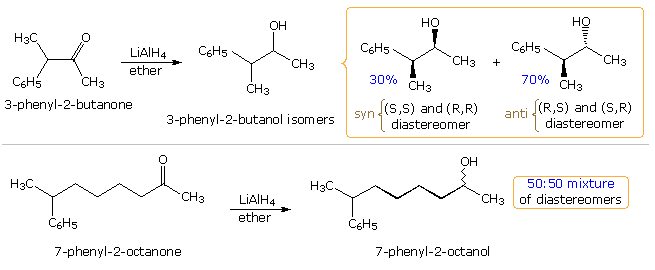
As expected, the behavior of conformationally mobile acyclic compounds is more difficult to rationalize. In the following illustration, 2 hydride reductions of chiral methyl ketones are shown. Since each ketone has an existing stereogenic site, and since the reduction creates a new chiral center, diastereomeric products are possible. These may be designated in several ways, but the syn-anti notation is generally preferred over the more express erythro-threo designation. When the existing stereogenic center is located adjacent to the carbonyl grouping, equally in the upper equation, it may influence the proportion of production diastereomers to a pregnant degree. Because the new stereogenic centers are vicinal, this is termed 1,ii-diastereoselectivity, and is the same for both an enantiomerically pure or a racemic reactant. If, however, the stereogenic eye is far abroad from the carbonyl group, it has a negligible influence on the reduction, and a nearly 50:50 mixture of diastereomers is produced (lower instance).
A number of models have been proposed to explain the diastereoselectivity of the first reduction. Many conformations nearly the alpha-C-CO bail may be written, and the claiming is to choice one that accounts for the observed selectivity. Three models will exist considered here. For general reference, the substituents on the chiral center next to the carbonyl group are labeled L (big), M (medium) and S (small), reflecting their guess size. The original construction of these models causeless an orthogonal (90º) approach of the nucleophile to the aeroplane of the carbonyl group, and a reactant-like transition state. In the following diagram this has been changed to the preferred Bürgi-Dunitz approach, every bit shown past a pinkish pointer. Each model predicts the right configuration of the favored diastereomer from LiAlH4 reduction of 3-phenyl-2-butanone (upper equation above with Fifty=Chalf dozenH5, M=CH3 & Due south=H). This diastereoselectivity is commonly termed Cram or Felkin selectivity.
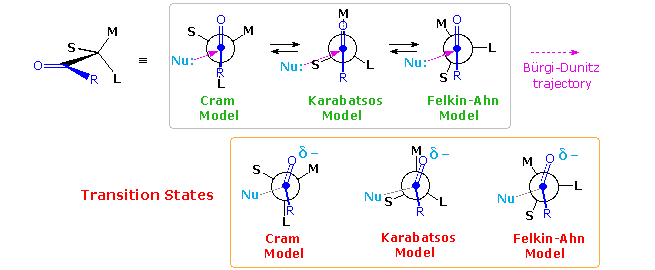
D. J. Cram (Nobel prize 1987) proposed the model on the left in 1952. Information technology suffers from steric hindrance of substituent L with the R substituent on the carbonyl group every bit well equally torsional strain. A more favored conformation was chosen by G. Karabatsos, equally shown by the center model. Hither the torsional strain is reduced, and the selectivity in reactions of phenylacetaldehydes bearing dissimilar α-substituents was rationalized more correctly than by Cram'due south model. The most contempo model (on the right) is that proposed by H. Felkin and elaborated by North.T. Ahn. In this case, overlap of the carbonyl π*-orbital with the C–50 σ*-orbital provides electronic stabilization. These models all require nomenclature of S, M & L substituents, occasionally a tricky process, and assume a reactant-like transition land for the reduction. Nonetheless, some bonding of hydride to the carbonyl carbon must have place in the transition state, accompanied by corresponding small structural changes. Plausible structures for these reactant-like transition states are given in the orange box.
A brief assay of these models is instructive. Although the unfavorable eclipsing of R & L iin the Cram model s slightly relieved in the transition state, the oxygen (and associated metal) moves toward the M group, resulting in increased steric crowding in that location. The Karabatsos model starts with a favorable conformer, but the nucleophile trajectory about eclipses the C-Southward bail (~20º dihedral angle). The oxygen shift in the transition state relieves eclipsing strain with G, benefiting the transition state energy. Finally, the Felkin-Ahn model seems to offering the all-time rationalization. The nucleophile trajectory is roughly 40º away from eclipsing the C-S bond, and crowding of both the O⇔M and R⇔Southward groups is reduced in the transition state.
Past clicking on the higher up diagram a tabular array giving the diastereoselectivity for seven typical addition reactions will announced. The excess of ane diastereomer over another is sometimes given as a ratio (e.g. 4:1), but here we employ pct diastereomeric excess (%de), divers equally the mole fraction difference in diastereomers times 100. Thus, a mixture of fourscore% diastereomer A and xx% diastereomer B (4:1) has a threescore% de. Beneath this tabular array is an instance of borohydride reduction of a steroidal methyl ketone (only ii rings are shown). The Felkin-Ahn model shown to the right of the equation correctly predicts the diastereoselectivity of the reduction in favor of isomer A. Remarkably, if this ketone is reduced by the bulky borane reagent, disiamylborane (CvH11)iiBH, the diastereoselectivity is reversed, with isomer B beingness formed in 82% de. Since this reduction gain by an initial complexing of the Lewis-acidic boron with the carbonyl oxygen, the favored transition country conformer is that in which the small hydrogen and the medium-sized carbon-16 methylene group exchange locations, influenced by the greatly increased size of the oxygen moiety. This change in stereoselectivity is sometimes termed anti-Cram or anti-Felkin selectivity. Aldehydes are especially prone to changes in stereoselectivity for the aforementioned reason; however, large nucleophilic reagents favor the Felkin-Ahn product due to reduced hindrance for the Bürgi-Dunitz trajectory (compare cases 1 & 2 in the table).
Because of the conformational mobility of these compounds, information technology is important to recognize an important precept known equally The Curtin-Hammett Principle.
The ratio of products obtained from a a group of equilibrating conformers is determined by transition state energies, not conformer concentrations.
The Chelation Effect
Shortly after proposing his initial model, Cram recognized that ketones and aldehydes having an α-oxygen, nitrogen or sulfur functional group often exhibited a different stereoselectivity. He attributed this beliefs to a cyclic chelated conformer, in which the hetero atom of the α-function is coordinated with the metal moiety of the nucleophilic reagent (e.g. Li. Al, B & Mg). The following diagram shows a typical reaction of this kind, in which the chelating ligand is Y and the proposed transition land is enclosed in square brackets. The nucleophile is shown attacking the carbonyl group preferentially from the side of the smallest remaining substituent. 9 examples are listed in the tabular array below the equation and two more are shown in the bluish tinted box to its right. Powerful coordinating metals such as Zn(Two) and Ti(IV) provide the strongest chelation control, equally does reaction at a depression temperature.
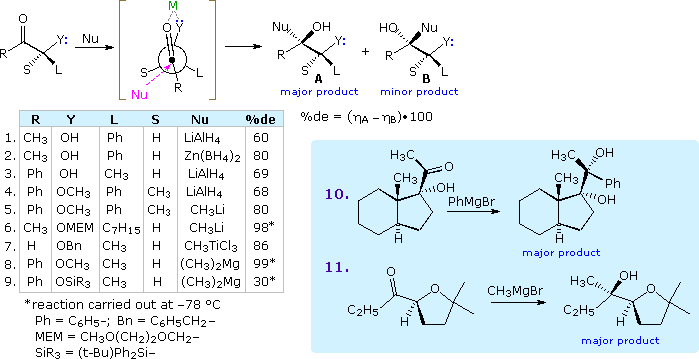
The silyl ether derivative in example 9 is a case of steric hindrance to chelation. The smaller trimethylsilyl ether derivative reacts chop-chop and with very loftier diastereoselectivity, whereas the larger triisopropylsilyl analog reacts slowly and with poor diastereoselectivity favoring isomer B.
Non-chelating Polar Effects
The influence of non-chelating polar substituents on the diastereoselectivity of carbonyl addition reactions proved dissonant. In a study of LiAlH4 reduction of 2-chloropropiophenone J. Cornforth noted that the syn-isomer B was formed in 3:1 ratio with its anti-isomer A, every bit shown by the superlative equation in the following diagram. Indications of substituent size from conformational equilibria of substituted cyclohexanes, suggest that a methyl group is significantly larger than a chlorine; consequently, the Felkin model predicts that the anti-isomer should predominate (xanthous shaded box). The chelation model leads to a similar prediction. Since chlorine is known to be a poor chelating ligand, Cornforth suggested that dipole repulsion with the carbonyl grouping (orange arrows) would result in an eclipsed conformation, such as that shown beneath the equation. Such an intermediate predicts the correct diastereoselectivity, merely information technology suffers from the same torsional strain equally the Cram model. The Felkin-Ahn model may be modified to reverberate the influence of a polar substituent, as shown by the formula right of center. This model as well predicts the correct diastereoselectivity, and suggests that boosted transition state stabilization may occur past overlap of the budgeted nucleophile with the antibonding σ* orbital of the C–Cl bond.
A further test of this rationalization is provided past removing the chelating metal species. Reduction of similar α-substituted propiophenones, C6H5COCHYCH3 (Y = dimethylamino or acetoxy), by the hypervalent hydride reagent, Chalf-dozenH5(CH3)2SiFH(–) (CfourH9)4N(+), gain with high diastereoselectivity, favoring the syn isomer analogous to B. The absenteeism of a chelating metal combined with the bulk of the hydride donor results in a >95 %de, despite the replacement of chlorine by the much stronger chelating ligands (CH3)2N- and CH3COtwo-.
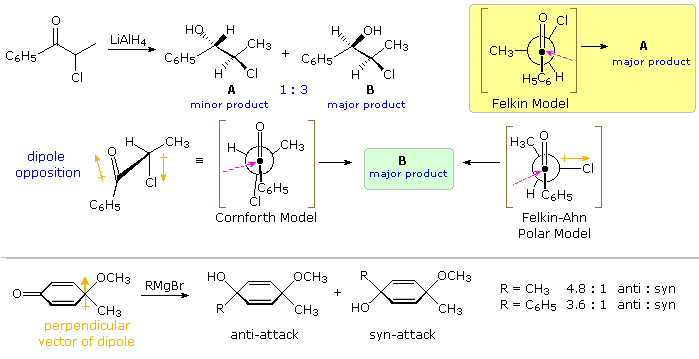
An interesting example in which steric effects and chelation are eliminated is shown at the bottom of the preceding diagram. Here a doubly vinylagous α-substituted ketone undergoes a diastereoselective addition of a Grignard reagent. This and related work by P. Wipf signal that the selectivity is proportional to the perpendicular component of the molecular dipole, with nucleophile approach favored from the positive side. Curiously, hydride addition occurs without any diastereoselectivity.
1:3-Diastereoselectivity
The previous discussion has focussed on examples of 1:2-diastereoselectivity, where the site of chiral influence has been side by side to the carbonyl function. If this influence is moved to the β-carbon its strongest effect is transmitted via conformers having six-membered chelate rings. Three examples of such 1:3-diastereoselectivity are shown in the following diagram. For the addition of organometallic nucleophiles a strong coordinating metal similar Ti(IV) is needed, both to stabilize the chelate ring and to activate the carbonyl grouping. As noted in equation ii, the magnesium of a Grignard reagent does not serve this purpose fairly. A possible transition state for the titanium chelated reaction is drawn in the left green shaded box (notation the favored centric approach of the nucleophile). Alternatively, intramolecular control may provide selectivity, as shown in equation iii. Sodium triacetoxyborohydride is a weak hydride donor that is commonly used in weakly acidic solutions for reactions such every bit reductive amination; information technology does not reduce isolated ketones. Its use in the reduction of ii,6-dimethyl-5-hydroxy-three-heptanone produces excellent diastereoselectivity, as a issue of the intramolecular hydride transfer intermediate fatigued in the right green shaded box (the bulky R substituent prefers to occupy an equatorial-like position).
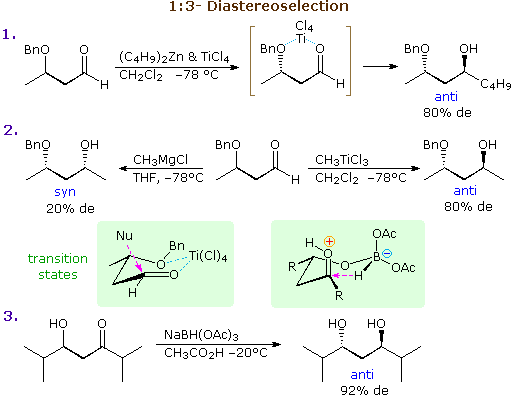
These same factors may also lead to 1:two-diastereoselectivity if α-substitution is nowadays in the substrate. By clicking on the above diagram four examples will be displayed there. In the first ii cases the α-carbon is the merely stereogenic heart, just selectivity occurs because of chelation to a β-functional group. Equations 6 & vii are boosted examples of steric control by intramolecular hydride transfer (see equation 3). Annotation that the big isopropyl groups control the facial selectivity, so the configuration of the smaller α-methyl substituent is relatively unimportant.
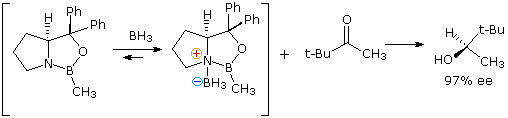
The intramolecular hydride transfer mechanism noted higher up serves as a model for achieving enantioselective reduction. Several novel catalysts, in which borohydride is complexed with a difunctional chiral ligand, have been developed and used for the enantioselective reduction of prochiral ketones to chiral alcohols. In the following example a chiral ligand prepared from proline is converted to a boron heterocycle. This goad binds reversibly with diborane to form the reactive reducing species. Coordination of the ketone oxygen with the Lewis acidic boron orients, and activates the carbonyl group for hydride transfer to its si-face.
Allyl and Crotyl Addition to Aldehydes and Ketones
Allyl Grignard reagents oft exhibit dissimilar reactivity patterns than like alkyl reagents, peculiarly with hindered ketones. For example, allylmagnesium bromide adds to diisopropyl ketone as expected, but the major product from reaction with north-propylmagnesium bromide is 2,4-dimethyl-3-pentanol, the upshot of reduction by a MVP mechanism. Reagents such as methylmagnesium chloride, which take no β-hydrogens to transfer, may act every bit strong bases and enolize hindered ketones having α-hydrogens, even though the allyl reagent adds in good yield. This characteristic behavior has been attributed to the conjugated (or ambident) nature of this carbon nucleophile, a feature that is readily demonstrated in crotyl (ii-butenyl) homologs.
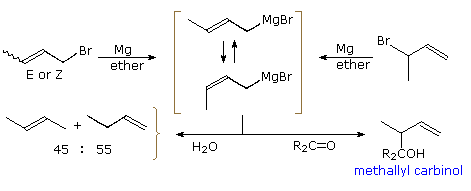
The chemistry of crotyl organometallic reagents has proven both interesting and useful. Conversion of either i-bromo-ii-butene (Due east or Z), or 3-bromo-ane-butene, to a Grignard reagent gives identical mixtures of equilibrating stereo and regioisomers. From nmr evidence, the 1º-isomers drawn within brackets in the following diagram, are the main components (the pocket-sized 2º-regioisomers are not fatigued). Rapid quenching of this Grignard reagent with water produces a most 50:l mixture of 1- and ii-butene (cis/trans mixture), suggesting facile protonation at both the 1º and 2º allylic sites. Remarkably, simple ketones and aldehydes react exclusively at the 2º-site to grade methallyl carbinols. A cyclic transition land for this rearranged addition will be displayed by clicking on the diagram. If the carbonyl group has different substituents, as shown, two new chiral centers are created (red asterisk), and a pair of racemic diastereomers will be formed. Accomplishing this C–C bond formation with high diastereoselectivity would provide chemists with a powerful tool for stereoselective synthesis of complex molecules. Such selectivity would also be unlike from most of the previous cases, in which diastereoselectivity results from the influence of a preexisting chiral center.
It should be noted that crotyl Grignard additions to very hindered ketones are reversible. The rearranged methallyl carbinol is the kinetically favored product, but estrus and extended reaction time leads to germination of the more stable crotyl carbinol, as illustrated past the lower equation in which the magnesium common salt of a sterically congested methallyl carbinol is the starting signal.
By clicking a second time on the above diagram, some examples of crotyl Grignard addition reactions volition be displayed. The greatest product diastereoselectivity is establish when i of the carbonyl substituents is much larger than the other. Because both E and Z-isomers of the crotyl reagent are present in the reaction, it is tempting to attribute the formation of the anti-diastereomer to i and the syn-diastereomer to the other. Two such transition states are drawn in the orangish box, the Due east-reagent country leading to the anti-diastereomer and the Z-reagent state to the syn-isomer. At this phase it is not wise to draw such a determination, since there are other plausible structures leading to both isomeric products, and according to the Curtin-Hammett principle the favored reaction path amidst competing possibilities will be that having the lowest activation energy. 1 additional case will be shown by clicking on the diagram a 3rd time. The steroidal 11-ketone in this example can only react with nucleophiles at its α or ri-confront, equally a consequence of hindrance by the β-oriented athwart methyl groups. Furthermore, the virtually favored approach of a crotyl moiety appears to be that of the Z-isomer shown in the orangish box. Indeed, a unmarried product having the (11R, 20R)-configuration shown was obtained and identified by 10-ray analysis. In this case the Z-crotyl reagent produces the anti-diastereomer.
 Considering of its potential usefulness in synthesis, organic chemists have sought ways to achieve predictable control of diastereoselectivity in the addition of methallyl groups to carbonyl compounds via crotyl reagents. To this cease a variety of other metals, ranging from zinc, chromium and titanium to boron, silicon and can, have been investigated. The zinc, titanium and chromium reagents tend to undergo rapid E/Z interconversion, and either react with mixed diastereoselection or with a strong bias toward anti-selectivity. The examples shown on the right for crotyltitanium triphenoxide illustrate the latter beliefs. Similarly, chromium (2) reagents generated in situ from (East or Z)-1-iodo-2-butene add together to benzaldehyde with loftier selectivity favoring the anti-diastereomer.
Considering of its potential usefulness in synthesis, organic chemists have sought ways to achieve predictable control of diastereoselectivity in the addition of methallyl groups to carbonyl compounds via crotyl reagents. To this cease a variety of other metals, ranging from zinc, chromium and titanium to boron, silicon and can, have been investigated. The zinc, titanium and chromium reagents tend to undergo rapid E/Z interconversion, and either react with mixed diastereoselection or with a strong bias toward anti-selectivity. The examples shown on the right for crotyltitanium triphenoxide illustrate the latter beliefs. Similarly, chromium (2) reagents generated in situ from (East or Z)-1-iodo-2-butene add together to benzaldehyde with loftier selectivity favoring the anti-diastereomer.
Some chair-like cyclic transition states that accept been proposed for these reactions will exist displayed on the right by clicking on the diagram. The Eastward-based transition land A shown at the tiptop is presumed to exist favored, and leads to the anti-production isomer. An alternative Z-transition state, B would class the syn-isomer. The anti-isomer could also be formed by way of the Z-transition country D, drawn in the light grayness box. As noted before, the assignment of ketone substituents equally big, L and small, Southward, may sometimes be arguable.
Depending on what other ligands are present, crotyl boron, silicon and tin reagents have greater configurational stability than the metal reagents discussed above. Allyl boron and tin reagents may add together spontaneously to aldehydes, but allyl silanes mostly require catalysis by Lewis acids (this activates the carbonyl function) or past fluoride anions (this increases the nucleophilicity of the allyl moiety).
Addition reactions of a crotylstannane reagent are shown in the following diagram and illustrate some of import features. The E and Z isomers of the reagent are relatively stable at room temperature, but react sluggishly with virtually aldehydes. Chloral, existence more than electrophilic than nigh aldehydes, provides a convenient substrate for the uncatalyzed, room temperature improver of this reagent. The stereoselective reactions presented at the tiptop of the diagram advise a cyclic transition state machinery in which the E-crotyl reagent gives the anti-diastereomer, and the Z-reagent forms the syn-diastereomer.
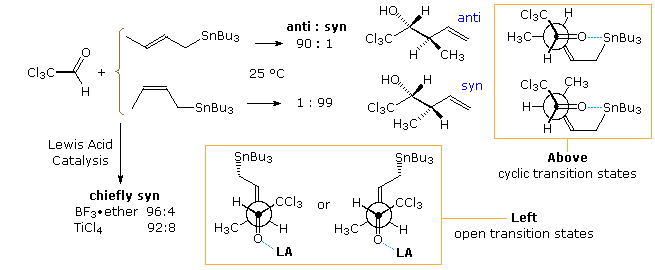
By an initial coordination of the carbonyl oxygen with a Lewis acid, the add-on of crotylstannane is accelerated, but both E and Z-isomers form the syn-diastereomer every bit the major production. Open or acyclic transition states, such as the 2 drawn at the lesser of the diagram, appear to be functioning in this modification of the reaction. Note that the deviation between these East and Z-transition states is very small, and the C–Sn bond is conjugated with the π-orbital of the double bail. Interestingly, if TiCliv is mixed with the crotyl stannane earlier it is added to the aldehyde (contrary add-on), the anti-isomer of the product is formed preferentially from both E and Z-reagents. A metal exchange reaction, in which the crotylstannane is converted to a crotyltitanium reagent about likely takes place. Equally noted above, the titanium reagents generally give anti-addition products. Lewis acid catalysis of crotyl silane addition reactions besides gain by manner of an open transition state, and favors the syn-production isomer.
Crotyl Boron Reagents
Although information technology is possible to prepare E and Z-crotylboranes, CHiiiCH=CHCH2B(R)two, they interconvert at room temperature. Consequently, any study of improver stereoselectivity for the individual isomers must exist conducted at -78 ºC. When this is done the Eastward-isomer favors the anti-product diastereomer, and the Z-isomer favors the syn-product, both with >98% de.
A more convenient approach is to use crotylboronate esters, CHthreeCH=CHCHiiB(OR)2, or crotyltrifluoroborate salts, CH3CH=CHCHiiBF3 (-) K(+), as the addition reagent. The stereoisomers of these compounds are not only configurationally stable at room temperature, only the compounds themselves are tolerant of exposure to air and moisture, making their grooming storage and utilise relatively elementary. An application using the pinacol ester of crotylboronic acid in shown in the following diagram. The presumed chair-like cyclic transition states for the addition are fatigued on the meridian, with the product from each given underneath. Newman projection drawings are shown in the orange box at the bottom. The data in the table demonstrates the excellent diastereoselection achieved in this reaction.
A like set of equations and data for crotyltrifluoroborate reactions will exist displayed in a higher place by clicking on the diagram. In this reaction addition is catalyzed by BF3-etherate, which functions as a fluoride ion transfer agent.
Enantioselectivity
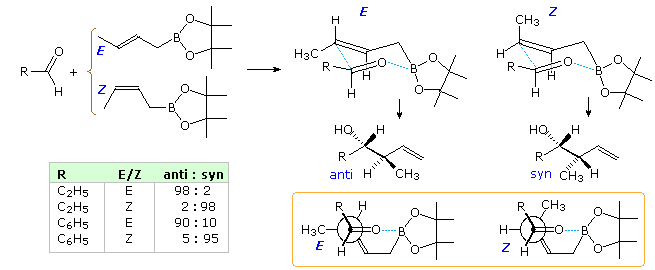
When achiral aldehydes and ketones are substrates for addition of allylic reagents, reaction takes place equally at both prochiral faces of the carbonyl double bond. For these improver reactions to be made enantioselective, the rate of reaction at i face of the double bond must be increased over that at the other face. This is the same requirement presented earlier for enantioselective hydroboration, and the isopinocampheyl chiral substituent used by Brown successfully in that example has proven effective here besides. Chiral boronate esters derived from tartrate esters have also served for enantioselective synthesis. Examples of both reagents are given in the following diagram. The specific chiral ligands indicated by the BLtwo* grouping are shown in the light green box at the upper correct of the diagram. Since both α-pinene and tartaric acrid enantiomers (antipodes) are available, the enantioselectivity of these reactions are easily controlled. The top equation shows enantioselective allyl add-on to a prochiral aldehyde, creating a single new stereogenic center. The bottom equation shows the analogous crotyl addition in which 2 new stereogenic centers are formed.
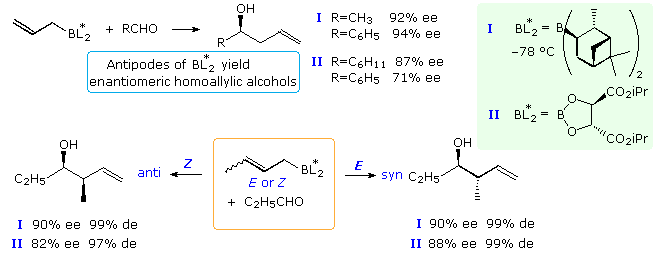
Crotylation of Chiral Aldehydes
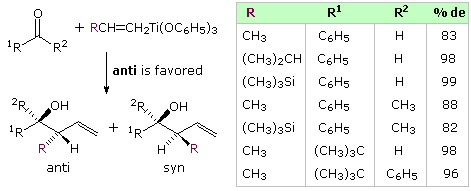
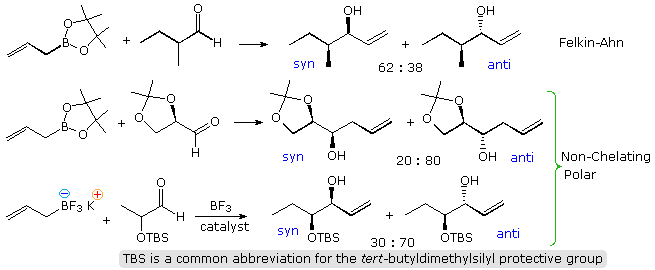
The beingness of a chiral center blastoff to a carbonyl group presents a second, potentially conflicting, cistron influencing the stereoselectivity of crotyl addition. The following diagram shows the improver of some achiral allyl boron reagents to three aldehyde substrates of this kind. The start instance may be rationalized by the Felkin-Ahn model, the poor stereoselectivity reflecting the similar size of methyl and ethyl groups. The other 2 examples have polar substituents at the α-carbon, and must be analyzed differently. Because the allyl add-on gain past a cyclic transition state incorporating a tetra-coordinate boron atom, the chelation model is not an option, and a non-chelation transition state all-around the polarity of the substituent provides the all-time caption. Only modest stereoselectivity takes place.
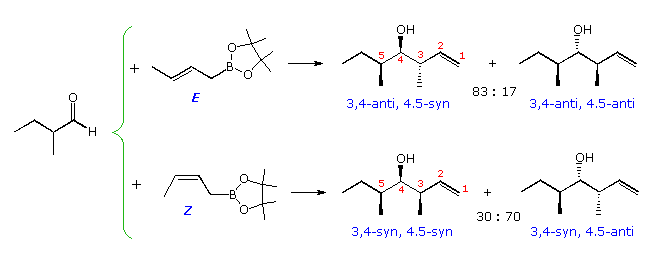
Analogous crotyl additions (both Due east and Z) have been reported for these aldehyde substrates. The reactions of 2-methylpropanal are shown in the diagram beneath. The slight preference for the syn-production noted higher up volition be reflected in the four,v-configuration of the products. The iii,4-configuration is generated by the chair-like circadian transition states described above, with E-crotyl reagents forming anti diastereomers and Z-reagents giving syn-diastereomers. This selectivity is conspicuously maintained in all the products, whereas the Felkin-Ahn control of 4,5-configuration is marginal. Fortuitously, the favored transition state for the E-reagent has a Felkin-Ahn configuration, but the Z-transition state does not.
Past clicking on the diagram favorable representations of the cyclic transition states for these reactions will exist displayed (R=CtwoH5). Newman projection views of the same transition states are drawn in the orange box.
The results of crotyl add-on reactions to the two aldehydes having polar α-substituents will exist displayed higher up by clicking a second and third time on the diagram. The dominant stereoselectivity results over again from the tendency of Eastward-crotyl reagents to grade anti diastereomers and Z-reagents syn-diastereomers. In both cases this is reflected by the iii,4-configuration (note the numbering alter for the glyceraldehyde acetal in case ii). The influence of the α-polar substituent is once again less meaning, and in these cases reinforcement takes place for the Z-reagent relative to its E-isomer.
The Aldol Reaction
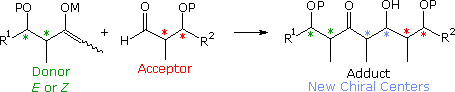
Improver reactions of enolate species to aldehydes and ketones, known as the aldol reaction, are structurally analogous to the allyl and crotyl addition reaction described higher up. This similarity, which is shown in the following diagram, extends to the creation of two new stereogenic centers, cherry asterisks, from accordingly substituted allyl and enolate reactants (R1 ≠ H). By varying the metal Grand from Li and Na through Mg, Zn and Ti to B and Si, its influence on the diastereoselectivity of these reactions has proven to be integral, with boron providing some of the best selectivity. It is appropriate and instructive, therefore, to examine how far the previous analyses and interpretations may be extended and applied to the aldol reaction.
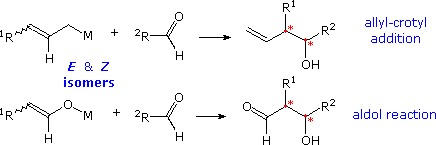
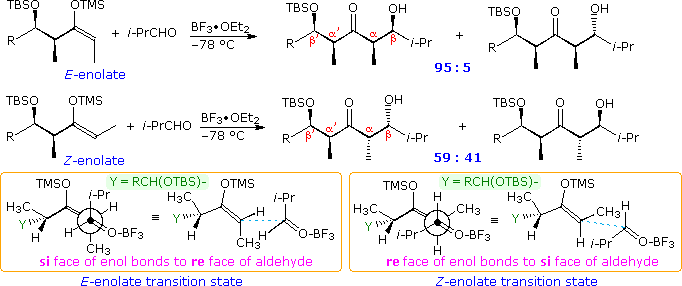
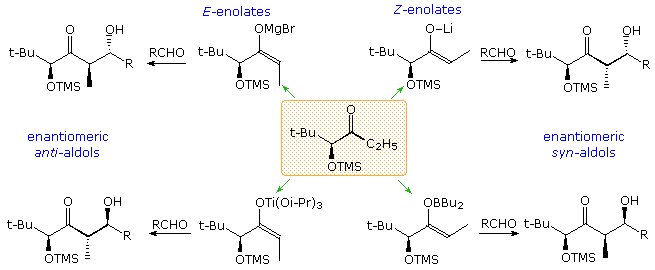
Our ability to command the donor-acceptor roles of carbonyl reactants, the regioselectivity and stereoselectivity of enolization, and the diastereoselectivity of the aldol product are discussed elsewhere in this text. Techniques for generating the E and Z isomers of designated enolate species take been developed, and the diastereoselectivity of each in the aldol reaction established. As a general rule for reactions involving circadian transition states, East-enolates produce anti-aldols, and Z-enolates the syn-diastereomer, a tendency that reflects the facial selectivities of the transition states. Thus the reaction of Due east-enolates with aldehydes proceeds preferentially by the re (or si) confront of the carbonyl reactant bonding to the same prochiral face of the enolate. Conversely, similar reactions of Z-enolates occur by preferential bonding of the re (or si) face of the aldehyde to the si (or re) face of the enolate. Enolborinates were among the virtually reliable and selective reagents revealed by a host of aldol studies, a quality reflected in reactions of the crotylboronates. The Mukaiyama aldol reaction of silyl enol ethers takes place by mode of an open transition state, in which both enolate configurations are biased to bond to the contrary prochiral face of the aldehyde, giving syn-diastereomers as the major product.
In light of previous discussions, it may be anticipated that the form of aldol reactions will exist farther influenced by the presence of a nearby chiral center in the carbonyl acceptor or the enolate donor. The following sections provide examples of such stereoselectivity.
i:2-Diastereoselection in Reactions with Chiral Aldehydes
Two aldol reactions of α-substituted phenylacetaldehydes are presented in the following diagram. The first is a typical brine metallic enolate add-on; the second, in which a silyl enol ether undergoes Lewis acid catalyzed addition, is called the Mukaiyama aldol. The attacking enolate anion is achiral, so the diastereoselectivity of the reaction is a measure out of the facial selectivity (re vs. si) imparted to the aldehyde carbonyl by the α-substituent. Because the α-substituent is an alkyl group in these reactions, the syn-anti classification for the product diastereomers is ambiguous, depending on which group (phenyl or alkyl) is considered a substituent. For comparison purposes in this drawing, the alkyl group is designated the substituent. Unexpectedly, as the alkyl substituent increases in size (equation ane), the moderate syn stereoselectivity is diminished and changes to a weak preference for the anti-isomer when R = tert-butyl. Four Felkin-Ahn models may be considered when analyzing these results. A and D predict syn-selectivity, while B and C pb to the anti-diastereomer. When R = methyl or ethyl, A is the favored transition land, but for the larger substituents, iso-propyl and tert-butyl, C becomes increasingly of import, tipping the diastereoselectivity toward the anti-product. Transition states B and D exhibit hindrance to nucleophile approach and are unlikely participants. A Zimmerman-Traxler cyclic transition state construction, analogous to A when R = CH3, is drawn in the orange shaded box. Equally R increases in size, its centric-like orientation suffers increased hindrance, favoring an culling state in which the R & Ph groups exchange locations (i.e. C).
The increased diastereoselectivity of the Mukaiyama aldol reaction is noteworthy (de is 92 versus 56 for the analogous instance in equation 1), and reflects the open up transition state adopted by this reaction. To visualize this state, rotate the aldehyde component 180º from its orientation in the cyclic structure, maintaining the developing carbon-carbon bond. This leaves the two oxygen functions far apart. Coordination of the aldehyde carbonyl with BF3 increases the electrophilicity of its carbon, and may serve to stabilize an orthogonal orientation of the adjacent phenyl group, as in A (and B). Such a configuration is known to be of import for anchimeric assist by an next phenyl group in carbocation formation and rearrangement.
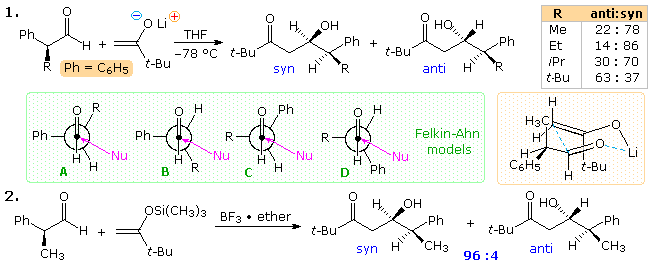
Aldol reactions of some chiral aldehydes having an α-heteroatom substituent will be displayed in a higher place by clicking on the diagram. Although these additions tend to favor the anti-diastereomer, steric factors often determine the caste of diastereoselectivity. The aforementioned ketone enolate used in the previous study is the nucleophile in reaction 3. When the R group is larger than methyl, skilful one,2-diastereoselectivity for the anti-isomer is observed. The Felkin-Ahn polar model serves to predict this outcome, as does a modification of the Cornforth model proposed by D.A. Evans, (Harvard). Reaction 4 makes utilize of a malonate nucleophile and, because of its greater stability and reduced reactivity, Lewis acrid activation of the aldehyde is required. Since both BF3 and ZnCl2 produce strong anti-diastereoselectivity, the transition state does not seem to be chelated. An increment in the size of the oxygen substituent P causes a decrease in diastereoselectivity, a trend that is all-time explained by the Evans model. In this respect, the very big trityl group induces a dramatic modify in diastereoselectivity, which might exist explained by the modified Evans model fatigued at the lower right.
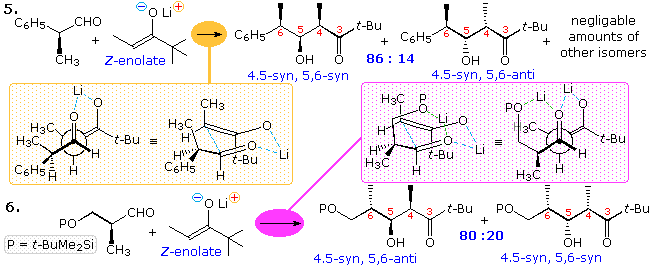
Two additional examples in which a Z-enolate is used as the nucleophile are drawn in the following diagram. Reaction 5 should exist compared with reaction ane of the before display. The very high 4,v-syn diastereoselectivity is typical of Z-enolates reacting via a cyclic transition state, equally drawn in the orange shaded box. The v,six-syn selectivity is slightly better than that for R = CHthree in equation 1, and conforms to the Felkin-Ahn model. Reaction 6 introduces the influence of an achiral β-siloxy part, capable of chelating with the carbonyl oxygen. The 4,5-diastereoselectivity remains exclusively syn, but the five,6-selectivity is changed to anti (60% de) as a event of chelation (magenta shaded box).
i:3-Diastereoselection in Reactions with Chiral Aldehydes
Two examples of 1:3-diastereoselection in reactions of β-substituted aldehydes are shown in the following diagram. Several limitations should be noted at the outset. Showtime, these are all Mukaiyama aldol reactions of silyl enol ethers. Since BF3 does not allow chelation, these reactions proceed by an open transition state. Similar reactions of enolborinate reactants, which involve a circadian transition state, take identify with very poor diastereoselectivity. Second, a β-alkyl substituent seems to have little influence, in contrast to the case of Y=CH3 in example ii above. All of the polar substituents except acetoxy display moderate to practiced diastereoselectivity in favor of the 1,three-anti diastereomer. A model for these reactions, in which steric and electrostatic repulsions are minimized, has been suggested past Evans. In this model the dipole of the polar Y substituent is oriented roughly anti to the carbonyl group, as shown at the lower left.
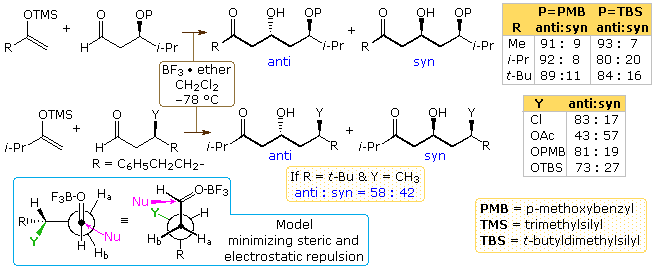
The add-on of an α-alkyl substituent to the aldehyde reactants used in the to a higher place written report creates a competition betwixt the syn-selectivity associated with the Felkin-Ahn model and the anti-selectivity noted here for polar β-substituents. Normally nosotros would expect the stereo center nearest the acceptor carbonyl function to exert a controlling influence; all the same, evidence to the opposite will be displayed above past clicking on the diagram. The anti-reactant isomer shown in the pinnacle equation adds with loftier diastereoselectivity favoring product isomer A. This diastereomer may be designated ane,2-syn,1,3-anti with reference to the newly formed chiral center (scarlet asterisk). A transition state for this add-on, fatigued at the lower left (gray shaded box), exhibits complementary features of both the Felkin-Ahn and electrostatic models. Equally a result of this stereoreinforcing influence, high diastereoselectivity is expected.
With the exception of enolates carrying the large tert-butyl group, the syn-reactant isomer adds with relatively poor diastereoselectivity. The R = tert-butyl enolate forms the 1,ii-syn,1,3-anti-diastereomer C with loftier selectivity, just the R = methyl enolate is much less selective and actually favors the 1,2-anti,1,3-anti-diastereomer D when P = PMB. Transition states leading to diastereomers C and D are drawn at the bottom of the diagram. In these cases the influence of the α and β- substituents conflict and are nonreinforcing. Felkin-Ahn dominance favors isomer C, whereas electrostatic factors favor formation of D. If the solvent is changed from methylene chloride to the less polar toluene, the diastereoselectivity shifts in favor of D in every example.
Information technology is interesting to encounter what happens to the selectivity described here when Due east and Z-enolate donors replace the methyl ketone enolates used above. Examples of these four reactant combinations are shown in the post-obit illustration. In all but the last Z-enolate reaction Felkin-Ahn facial selectivity dominates, with β:γ being syn in products A through D. Also the known preference of the Mukaiyama aldol for α:β syn products is realized with a de varying from 40% to 90%. Information technology should be noted that the anti-configuration of substituents in aldehyde I mutually reinforce Felkin-Ahn command, especially with the E-enolate. As noted above. the syn-substituents in Two are nonreinforcing, leading to a lower diastereoselectivity in the aldol products.
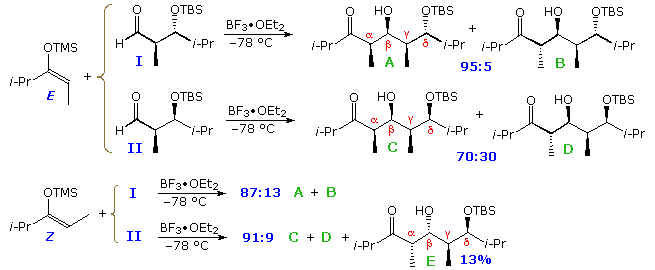
Corresponding reactions of aldehydes I and 2 (above) with comparable E-enolborinates and Z-chlorotitanium enolates are shown below. The circadian transition states of these reactions impose constraints that are evident in the product isomer distributions. As expected, the E-borinates give α:β-anti diastereomers exclusively, and the Z-titanium enolates strongly favor the α:β-syn family of isomers. Felkin-Ahn control is only stiff in the reaction of the mutually reinforced anti-substituted aldehyde I with the Due east-borinate. Other combinations show diminished β:γ-syn selectivity, and in the case of the Z-titanium enolates anti-Felkin-Ahn selectivity narrowly predominates. The facial bias imposed on the aldehyde carbonyl group by a β-polar substituent may be seen in the proportions of diastereomers having 1,three-anti (β:δ-anti) configurations (east.grand. A, B, East & G).
Diastereoselection in Reactions with Chiral Enolates
The enolate donor in an aldol reaction may likewise have a center of chirality, leading to the germination of additional diastereomeric products. The 2 equations in the following diagram evidence examples in which 1,4-diastereoselection (cerise asterisks) might result from such an aldol reaction involving enolate derivatives of a methyl ketone. Annotation that the enolate in equation 1 is racemic, whereas that in equation 2 is the S-enantiomer. The prove indicates mediocre selectivity, probably resulting from steric differences between large and pocket-sized substituents (RL and RDue south) at the chiral middle. Because of a tighter cyclic transition state, the diastereoselectivity of boron enolates is greater than that of their lithium counterparts.
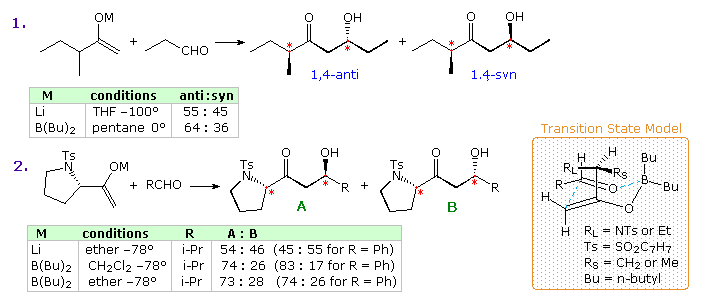
Two additional examples will be displayed to a higher place past clicking on the diagram. These Z-enolates are expected to favor 1,two-syn diastereoselectivity of the newly created α & β chiral centers, every bit noted earlier. The lithium enolate in example 3 exhibits moderate 1,2-syn selectivity, which is much improved when the corresponding enolborinate is used. Interestingly, the one,4-diastereoselectivity noted in case 2 (previous diagram) is enhanced for both enolate reactions, reflecting a preference for bonding from the si-face of the enolate species to the re-face of the carbonyl group every bit shown. Example 4 demonstrates the infrequent 1,ii- and one,iv-diastereoselectivity that can exist achieved with both enolborinates and chlorotitanium enolates. Notation that the selectivity given in the table is for the syn-syn diastereomer versus the other three isomers combined.
Three examples of reactions involving E-enolates are shown beneath. Fantabulous facial selectivity is establish in reactions of these nucleophiles Hither the 1,ii-diastereoselectivity of the newly created α & β chiral centers is strongly anti, equally expected. However, the 1,four-diastereoselectivity (α':β) is not consistent. The 1,four-anti-selectivity shown in reactions 6 and 7 is predicted by the transition state model, merely the ane,iv-syn-selectivity and one,3-anti selectivity (α':α) in reaction 5 is anomalous.
By clicking on the diagram, respective reactions of equivalent chlorotitanium Z-enolates will be shown. Again, strong facial selectivity is displayed for bonding at the re-face of the enolate as drawn, with both the new one,2- (α:β) and i,4- (α':β) diastereoselectivities being syn, as expected.

Analogous Mukaiyama aldol reactions are shown in the following diagram. The E- and Z-enolate reactants are both derived from the same syn-disubstituted ethyl ketone. Both enolates react with splendid but complementary facial selectivity. From past observations, the one,2-diastereoselectivity of the newly created α & β chiral centers is expected to exhibit moderate syn-diastereoselectivity. This is pronounced for the East- enolate, just very poor in the Z-isomer reaction. The exceptional and unusual 1,iii-anti selectivity (α':α) shown by the Z-enolate is noteworthy.
A final case of the remarkable directive influence that neighboring chiral centers may exert on carbon-carbon bond forming reactions is found in the 1,five-diastereoselectivity induced past β-substituents nowadays in methyl ketone donors. Examples are given in the following diagram, the 1,5-relationship being designated by the red asterisk in the top equation.
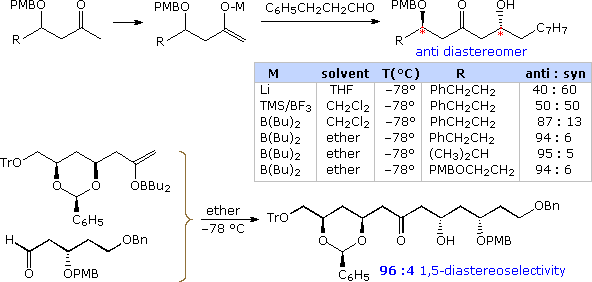
Supporting and Conflicting Substituent Effects
Many factors influence the diastereoselectivity of aldol reactions. These include the nature of the reaction (cyclic or open up transition state), the configuration of the enolate donor (Eastward or Z), the presence of stereogenic centers α to the carbonyl acceptor and/or the enolate donor, every bit well equally a β-polar substituent on the aldehyde. The following full general equation shows all these features present in one reaction (the colored asterisks designate chiral centers). Clearly, there are many possible perturbations of these factors, and establishing which combinations atomic number 82 to useful stereoselectivity is a challenge.
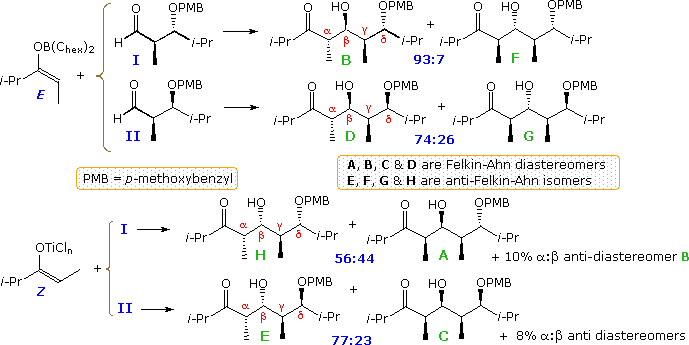
Since both reactants are chiral, it is desirable to design experiments that use enantiomerically pure reactants, so as to simplify the interpretation of results. Much of the work in this field has been carried out by the inquiry group of Prof. D. A. Evans (Harvard), the post-obit examples coming from their reports. The first examples. shown below, involve the E-borinate enolate from a syn-α-methyl, β-trimethylsiloxy-ethyl ketone reacting with syn and anti-α-methyl, β-alkoxyhexanals. The ketone enolate is a unmarried enantiomer; the aldehyde reactants in reactions A and D are enantiomers, as are the aldehydes in reactions B and C. The chiral centers from the aldehyde component are labeled α & β, the newly formed chiral centers are designated past lite blue asterisks, and the mutual centers from the enolate are labeled α' & β' (top equation). Past clicking on the diagram, corresponding reactions of the Z-titanium chloride enolate will exist displayed. Again, the aldehyde reactants in reactions A and D are enantiomers, equally are the aldehydes in reactions B and C; however, they have changed their location.
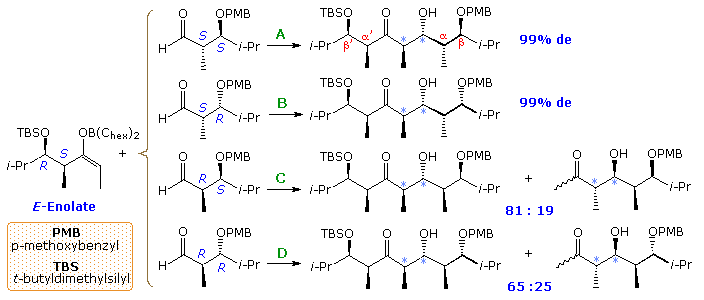
All the reactions of the E-enolate give anti-aldol products (calorie-free blue asterisks). In reaction A the α and β-stereogenic centers of the aldehyde and the α'-middle of the enolate accept matching influences on product diastereoselectivity. Thus Felkin-Ahn selectivity in which the re-face of the enolate bonds to the si-face of the carbonyl predominates. In reaction B the β-polar substituent of the aldehyde is mismatched in this respect, only has little effect on the overall diastereoselectivity. In reaction C the α-substituent of the aldehyde is mismatched, leading to dominance of the anti-Felkin-Ahn product. Finally, reaction D shows the results of a fully mismatched combination, which even produces a minor amount of syn-aldol product (non shown).
Nearly of the Z-enolate reactions shown by clicking on the diagram give syn-aldol products, as expected (light blue asterisks). Reaction A illustrates the fully matched influence of all the reactant stereogenic centers. Reactions B and C are the partially mismatched cases, and D shows a mixture of products from the fully mismatched combination. Reactions A and B display the anti-Felkin-Ahn preference associated with syn-aldol reactions. The remarkable influence of a β-polar substituent on the aldehyde is again shown in reaction C, where a strong shift to Felkin-Ahn improver occurs, despite the mismatched β-methyl substituent.
A similar study of the Mukaiyama aldol reaction is outlined in the following diagram. Since this reaction tends to give syn-aldol products, those combinations of reactants having an additional bias toward syn-selectivity have been selected. As shown, these four combinations do indeed exhibit a high degree of syn-diastereoselectivity (light bluish asterisks), and also follow Felkin-Ahn selectivity. Although the Z-enolate reactions are less selective, they produce a loftier degree of 1,3-anti-dimethyl isomers in dissimilarity to the corresponding syn-configurations obtained in other reactions.
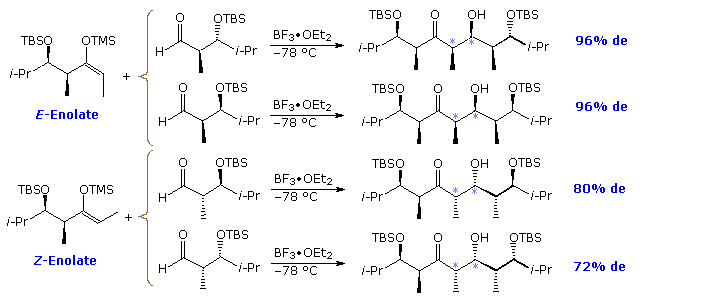
The remaining four reactant combinations, not shown, give mixtures of diastereomers that include pregnant amounts of anti-aldol products.
Enantioselective Aldol Reactions
Chiral Borinate Enols
Aldol reactions of prochiral donor and acceptor reactants produce racemic mixtures of chiral adducts. For such reactions to exist made enantioselective, it is necessary to introduce a chiral feature that volition induce a difference in reaction rates at the prochiral faces of the acceptor carbonyl role. This is the same requirement presented before for enantioselective crotylation reactions, and chiral borinate moieties, such as those shown in the post-obit diagram, accept been used for this purpose. One instance of this application is drawn below the formulas I through IV, and iv more volition be displayed by clicking on the diagram. Note that the last example shows the addition of an acetate unit of measurement, so enantioselectivity is non accompanied past diastereoselectivity.
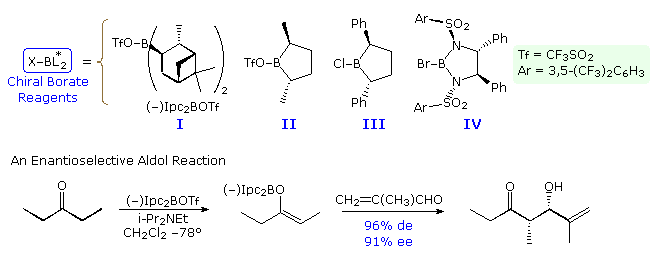
A model of the transition state for the isopinocampheyl instance may exist examined by .
Chiral Auxiliaries
Another approach to enantioselective aldol synthesis requires attachment of an enantiomerically pure chiral substituent to one of the reactants in the reaction. If this substituent exerts a controlling influence, and if the diastereoselectivity of the reaction is excellent, the product should be obtained as a single enantiomer. The chiral substituent may so be removed, yielding the final enantiomerically pure aldol adduct. A substituent serving this purpose is commonly called a chiral auxiliary. Chiral auxiliaries take been prepared from dissimilar kinds of natural products, including amino acids, alkaloids and terpenes. In this section of the text we shall make use of a family of oxazolidinone auxiliaries prepared by the Evans group (Harvard). Three examples of these auxiliaries are shown in the post-obit diagram. Many other types accept been prepared and used in a variety of reactions. The oxazolidinone auxiliary is usually incorporated as an imide derivative, shown for propionic acrid in the equation at the bottom of the illustration. A Z-enolborinate is prepared in the usual mode, and this reacts with a number of achiral aldehydes with very high enantioselectivity. In this instance the re-face of the enolate bonds to the si-confront of the aldehyde.
Although the enolborinate past itself might be expected to exist in a chelated form, with two B–O bonds, the aldol reaction requires a reorganization of this chelation in order to actuate the aldehyde carbonyl group for nucleophilic add-on. Equally shown by the formula in brackets, the costless oxazolidinone band has rotated 180º from its chelated position in order to minimize dipole repulsion. Steric hindrance by the pendent isopropyl grouping directs the reaction to the 2Due south,3R production.
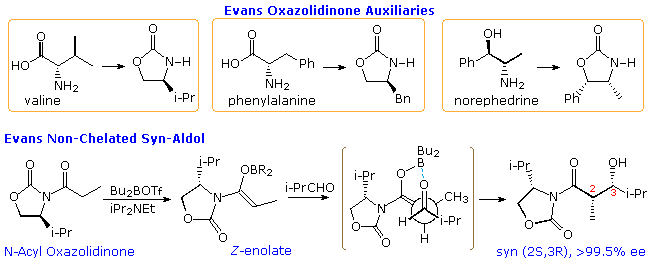
By clicking on the diagram, two additional examples of the "Evans aldol" will be displayed. Note that the configurational difference between the valine and norephedrine derived auxiliaries leads to unlike facial selectivity in the reaction, thus yielding the other syn-enantiomer in pure form. The chiral auxiliary group may be removed to give either the carboxylic acid or its methyl ester by base of operations catalyzed cleavage. Racemization of the α-carbon is possible, but seems to exist negligible. Other procedures for removing the auxiliary have as well been developed, with lithium hydroperoxide beingness particularly selective thanks to the alpha issue. In all these cases no anti-diastereomers are formed.
If the auxiliary remains chelated to the enolate during the aldol reaction the stereochemical outcome is inverse. Past clicking on the diagram a second time , two examples of this phenomenon will be shown above. In the upper equation a chelated Z-titanium enolate is initially formed and so reacted with an aldehyde. In contrast to boron, the ligand beat out of titanium is readily increases, permitting the activated complex (in brackets) to maintain the chiral auxiliary in a chelated orientation. The steric influence of the auxiliary is therefore opposite to that exerted in the unchelated borinate reaction, and the major product is the 2R,3South-syn-enantiomer, South1. Small amounts of the syn-enantiomer, Stwo, and anti-diastereomer A1 are also formed. The lower equation describes a unlike procedure which leads to the same major product. Here, a complexed Z-enolborinate is prepared and then allowed to react with 2 equivalents of aldehyde activated by complexation with a Lewis acrid such as TiCliv or SnCl4. An open transition land, like to that of the Mukaiyama aldol reaction, has been proposed by C. H. Heathcock (Berkeley) and is drawn in brackets. The simply minor isomer obtained was Aane. Interestingly, when the big Lewis acid (C2H5)2AlCl was used to activate the aldehyde, the anti-enantiomer Aane was formed in xc% de, accompanied past S2. Steric hindrance of the Lewis acid with the Z-methyl group changes the facial selectivity of the aldehyde from re to si.
As a rule, Evans' chiral auxiliaries exert a controlling influence in reactions with chiral α-substituted aldehydes, overriding even Felkin-Ahn preferences.
Selective Enolization of an α-Substituted Chiral Donor
Statistically, the reaction of a prochiral enolate with a prochiral aldehyde is probable to produce a mixture of 4 diastereomeric aldols as their racemates. In seeking to directly such reactions to a single stereoisomer the post-obit features must exist controlled.
• The enolate configuration E or Z.
• The facial selectivity of the enolate donor.
• The facial selectivity of the aldehyde acceptor.
If an α-substituent renders the enolate moiety chiral, and it is used as a single enantiomer, diastereomeric control of the factors listed above would lead to the formation of a single stereoisomer. This outcome has been realized in a remarkable study by C.H. Heathcock (Berkeley), in which all four possible aldol diastereomers were selectively prepared from the reaction of (Southward)-4-trimethylsiloxy-5,v-dimethyl-3-hexanone with an assortment of aldehydes, equally summarized in the following diagram. The starting ketone is drawn in the center orangish shaded box, and the preparation of each syn and anti-enantiomer was achieved past selective enolate formation, equally designated by the green arrows.
By clicking on the diagram, the two procedures leading to the syn-enantiomers will be displayed. The change in selectivity relative to the siloxy substituent is due to its chelation outcome in the lithium enolate and non-chelated polar effect in the boron enolate. As a issue, the beefy t-butyl grouping serves to direct bonding from the re-face of the enolate to the si-face up of the carbonyl group in the lithium enolate, but changes the facial selectivity of both reactants in the enol borinate.
Clicking on the diagram a second time changes the display to procedures leading to the anti-enantiomers. Pure E-enolates are more difficult to generate, and the discovery that the magnesium table salt from reaction of 2,2,6,six-tetramethylpiperidine with ethylmagnesium bromide enolized the starting ketone selectively in this manner was crucial. In the new display this enolization is described on the far left by way of the bracketed transition structure. The α-siloxy substituent over again chelates with the magnesium favoring a transition land in which the si-face of the enolate bonds to the si-face of the carbonyl group (top equation). In social club to reverse this selectivity, a bulkier silyl substituent is introduced and the magnesium is exchanged with a triisopropoxidetitanium moiety. This exchange required a specific combination of solvents and ultra-sound activation. In one case again, chelation is prevented, and dipole opposition causes a reversal in facial selectivities, leading to the enantiomeric anti-aldol product.
Culling routes to E-enolate intermediates are possible. Procedures for the grooming of E-enolborinates have been described, but in this example the α-siloxy substituent appears to interfere with this approach. Clearly, diastereoselective and enantioselective aldol synthesis requires careful evaluation of the many factors that may influence a specific awarding.
| Render to Table of Contents |
|---|
This page is the holding of William Reusch. Comments, questions and errors should be sent to whreusch@msu.edu.
These pages are provided to the IOCD to assist in capacity building in chemical education. 05/05/2013
![]()
Source: https://www2.chemistry.msu.edu/faculty/reusch/virttxtjml/sterslct.htm
0 Response to "How Do You Know if a Reaction Is Closed or Open Transition State"
Post a Comment